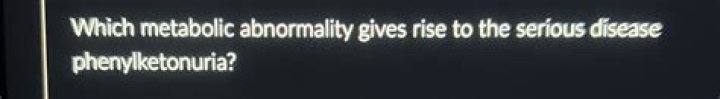
Which metabolic abnormality gives rise to the serious disease phenylketonuria
Phenylketonuria (fen-ul-key-toe-NU-ree-uh), also called PKU, is a rare inherited disorder
Which metabolic abnormality gives rise to phenylketonuria?
Mutations in the PAH gene cause phenylketonuria. The PAH gene provides instructions for making an enzyme called phenylalanine hydroxylase . This enzyme converts the amino acid phenylalanine to other important compounds in the body.
How does phenylketonuria affect metabolism?
Phenylketonuria (PKU) is an inborn error of phenylalanine (Phe) metabolism caused by the deficiency of phenylalanine hydroxylase. This deficiency leads to the accumulation of Phe and its metabolites in tissues and body fluids of PKU patients.
What is phenylketonuria metabolic?
Phenylketonuria (PKU) is an autosomal recessive inborn error of phenylalanine (Phe) metabolism resulting from deficiency of phenylalanine hydroxylase (PAH). Most forms of PKU and hyperphenylalaninaemia (HPA) are caused by mutations in the PAH gene on chromosome 12q23.Does phenylketonuria cause metabolic acidosis?
PhenylketonuriaFrequency~1 in 12,000 newborns
What kind of mutation is phenylketonuria?
Classical PKU is an autosomal recessive disorder, caused by mutations in both alleles of the gene for phenylalanine hydroxylase (PAH), found on chromosome 12.
Which enzyme is deficient in phenylketonuria?
PKU is characterized by absence or deficiency of an enzyme called phenylalanine hydroxylase (PAH), responsible for processing the amino acid phenylalanine. Amino acids are the chemical building blocks of proteins, and are essential for proper growth and development.
What is the biochemical basis of phenylketonuria?
Phenylketonuria is an autosomal recessive inherited disease caused by a disturbance in the phenylalanine hydroxylating system. Phenylalanine is converted to tyrosine by phenylalanine hydroxylase, which is located mainly in the liver. This enzyme needs the reduced cofactor tetrahydrobiopterin to be active.What is phenylketonuria Pubmed?
Phenylketonuria (PKU) is an autosomal recessive inborn error of metabolism caused by a deficiency in the hepatic enzyme phenylalanine hydroxylase (PAH). If left untreated, the main clinical feature is intellectual disability.
What cells are affected by phenylketonuria?PKU affects the brain. The signaling molecules that brain cells use to communicate with each other are called neurotransmitters. When neurotransmitters are not made in the right amounts, the brain cannot function properly.
Article first time published onWhat is the effect of the mutation on the cell tissue phenylketonuria?
Mutations in the PAH gene can cause phenylketonuria (PKU), a disorder that can change cells in the brain. The faulty protein allows dangerously high levels of phenylalanine to accumulate in the brain, poisoning the cells. If a person with PKU consumes too much phenylalanine, the build-up can cause mental retardation.
When does phenylketonuria develop?
Different forms of phenylketonuria vary in their severity of signs. Classic phenylketonuria (PKU) is the most severe form. Babies with PKU usually seem healthy at birth. Signs of PKU begin to appear around six months of age.
What is neonatal Tyrosinemia?
Transient tyrosinemia of the newborn is a benign disorder of tyrosine metabolism detected upon newborn screening and often observed in premature infants. It shows no clinical symptoms. It is characterized by tyrosinemia, moderate hyperphenylalaninemia, and tyrosiluria that usually resolve after 2 months of age.
What is the so called Guthrie test based on?
The Guthrie test, also called the PKU test, is a diagnostic tool to test infants for phenylketonuria a few days after birth. To administer the Guthrie test, doctors use Guthrie cards to collect capillary blood from an infant’s heel, and the cards are saved for later testing.
What type of enzyme is phenylalanine hydroxylase?
PAHshowRNA expression patternshowGene ontologyOrthologsSpeciesHumanMouse
What are the different types of PKU?
- Hyperphenylalaninemia: the lowest level above normal.
- Mild PKU: blood levels are mildly elevated.
- Moderate or variant: levels are not low but not high.
- Classic PKU: blood levels of phenelalanine are high.
What is classical phenylketonuria?
Classic phenylketonuria (PKU) is an inherited (genetic) condition that prevents the body from processing proteins correctly. Your body breaks down the protein that you eat into parts called amino acids. Your body then uses those amino acids to make other proteins that it needs to function.
How many PKU mutations are there?
More than 500 mutations in the PAH gene have been identified in people with phenylketonuria (PKU). Most of these mutations change single amino acids in phenylalanine hydroxylase.
Is phenylketonuria a substitution mutation?
An amino-acid substitution involved in phenylketonuria is in linkage disequilibrium with DNA haplotype 2. Nature.
Is phenylketonuria a point mutation?
Point mutations in the PAH gene are known to cause PKU in various ethnic groups, and large deletions or duplications account for up to 3% of the PAH mutations in some ethnic groups.
What does Hyperphenylalaninemia mean?
Hyperphenylalaninemia is broadly defined as the presence of blood phenylalanine levels that exceed the limits of the upper reference range (2 mg/dL or 120 µmol/L) without treatment but that are below the level found in patients with phenylketonuria (PKU).
How is phenylketonuria diagnosis?
PKU is diagnosed with a blood test. In the United States and most other countries, a blood test is taken through a heel stick on newborn babies within 48 hours of birth. Further tests will be required to confirm the type of PKU and plan the best way of treating it.
Which of the following is diagnostic of Tyrosinemia Type 1?
Tyrosinemia type ICausesGenetic (autosomal recessive)Diagnostic methodDried blood spot testing, urinalysis, genetic testing
What are the cardinal manifestations of PKU?
A musty odor in the breath, skin or urine, caused by too much phenylalanine in the body. Neurological problems that may include seizures. Skin rashes (eczema) Fair skin and blue eyes, because phenylalanine can’t transform into melanin — the pigment responsible for hair and skin tone.
What is the pathophysiology of phenylketonuria?
Phenylketonuria (PKU) is an inborn error of phenylalanine (Phe) metabolism caused by the deficiency of phenylalanine hydroxylase. This deficiency leads to the accumulation of Phe and its metabolites in tissues and body fluids of PKU patients.
What body parts does phenylketonuria affect?
It is needed to break down the essential amino acid phenylalanine. Phenylalanine is found in foods that contain protein. Without the enzyme, levels of phenylalanine build up in the body. This buildup can harm the central nervous system and cause brain damage.
What happens in the cells and in the body of a person with phenylketonuria?
The PKU gene tells the cell to make an enzyme that breaks down the amino acid phenylalanine. Faults in the genes (mutations) may cause problems in the body because the correct message is not being sent. In PKU, the cells are not making the enzyme that breaks down phenylalanine, so it builds up in the blood and tissues.
What is phenylalanine made from?
Good sources of phenylalanine are eggs, chicken, liver, beef, milk, and soybeans. Another common source of phenylalanine is anything sweetened with the artificial sweetener aspartame, such as diet drinks, diet foods and medication; the metabolism of aspartame produces phenylalanine as one of the compound’s metabolites.
Is phenylketonuria an example of pleiotropy?
One of the most widely cited examples of pleiotropy in humans is phenylketonuria (PKU). This disorder is caused by a deficiency of the enzyme phenylalanine hydroxylase, which is necessary to convert the essential amino acid phenylalanine to tyrosine.
What is elevated tyrosine?
Tyrosinemia is a genetic disorder characterized by disruptions in the multistep process that breaks down the amino acid tyrosine, a building block of most proteins. If untreated, tyrosine and its byproducts build up in tissues and organs, which can lead to serious health problems.
What does elevated tyrosine mean?
High amounts tyrosine in the blood and an amino acid called succinylacetone in the urine might indicate that your baby has TYR I. Sometimes follow-up testing may also include testing a very small sample of skin.